Introduction to Quantum Chemistry Simulations in Drug Discovery
What is Quantum Chemistry?
Quantum chemistry represents a pivotal branch of chemistry where quantum mechanics principles are harnessed to decode molecular structures, behaviors, and reactions. By examining atomic and molecular interactions at their most fundamental level, this discipline employs sophisticated mathematical frameworks to anticipate and elucidate chemical phenomena. Grasping these core interactions is indispensable for unlocking the secrets of chemical reactivity. Unlike classical mechanics, which falls short in atomic-scale explanations, quantum chemistry provides the necessary depth.
Central to this field is the Schrödinger equation, which delineates electron energy states and wave functions within atoms and molecules. Through these computations, chemists can forecast critical molecular attributes—bond distances, angles, and vibrational patterns—while shedding light on chemical bonding dynamics. This knowledge is instrumental in deciphering how atoms and molecules interact.
The Schrödinger Equation and its Significance
The Schrödinger equation stands as a cornerstone of quantum chemistry. This mathematical formulation tracks the evolution of a quantum system's state over time. When solved for a molecule, it reveals energy levels and wave functions, which are paramount for comprehending its structure and reactivity.
Mastering this equation enables predictions about electron behavior in molecules. Its solutions paint a detailed portrait of electron distribution, directly influencing molecular properties and reactivity. As a predictive tool, it is unmatched in forecasting reaction outcomes and molecular interactions.
Molecular Structure and Bonding
Quantum chemistry is indispensable for unraveling molecular architecture and chemical bonds. By leveraging quantum principles, scientists can anticipate atomic arrangements and bond types within molecules. Such insights are vital for understanding molecular properties and their reactive tendencies.
This knowledge of structure and bonding allows researchers to predict molecular interactions and reactions across diverse chemical environments. These predictive capabilities form the bedrock of innovation in materials science, catalysis, and pharmaceutical development.
Computational Quantum Chemistry
The field relies on cutting-edge computational tools to solve the Schrödinger equation for complex molecular systems. These advanced techniques facilitate the prediction of molecular properties and reaction pathways that elude experimental observation.
Such computational methods are particularly crucial for studying large, intricate molecules, offering researchers a window into systems that defy traditional experimental analysis. This computational approach has become an invaluable asset in tackling pressing challenges across chemical disciplines.
Applications of Quantum Chemistry
The reach of quantum chemistry extends across multiple scientific domains, from materials science to biochemistry. In materials development, it aids in crafting substances with tailored properties, while in catalysis, it illuminates reaction mechanisms and catalyst design.
Within biochemistry, it provides molecular-level insights into biological processes. These understandings are critical for drug development, enzyme mechanism elucidation, and fundamental biological discovery. As a rapidly advancing field, quantum chemistry continues to reshape our comprehension of the natural world.
Predicting Molecular Properties with Quantum Chemistry
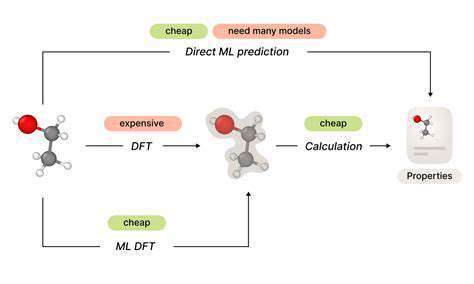
Predicting Molecular Properties Using Machine Learning
Machine learning has emerged as a transformative approach for molecular property prediction, offering distinct advantages over conventional experimental techniques, especially for complex systems or expansive datasets. The speed of machine learning models enables rapid property estimation, allowing scientists to navigate vast chemical landscapes and potentially uncover novel materials with targeted characteristics. These models learn from comprehensive datasets encompassing diverse molecular structures and their associated properties, identifying underlying patterns and correlations.
Additionally, machine learning excels at capturing complex structure-property relationships that challenge traditional methods. This capability proves particularly valuable when predicting properties such as reactivity, stability, and bioactivity. By processing enormous datasets, these models can detect subtle patterns that might escape human notice, yielding more precise and meaningful predictions.
Key Applications in Materials Science
Molecular property prediction holds revolutionary potential for materials science. Scientists leverage these predictions to engineer novel materials with customized properties, fast-tracking innovation in energy storage, catalysis, and electronic technologies. For example, predicting polymer mechanical properties prior to synthesis can dramatically streamline material development processes. This predictive approach enables a more focused and productive exploration of chemical possibilities.
The capability to forecast material performance before actual synthesis is transformative. It allows researchers to concentrate on the most promising candidates, conserving resources that might otherwise be wasted on less optimal options. This predictive power is revolutionizing material discovery and development across multiple industries.
Challenges and Future Directions
Despite its promise, machine learning for molecular property prediction faces several hurdles. A primary challenge involves securing high-quality, comprehensive training datasets. Inadequate or skewed data can produce unreliable predictions and constrain model generalizability. Another critical consideration is model interpretability. Grasping the rationale behind complex machine learning predictions is essential for establishing trust and ensuring their dependability.
Future research will target these challenges while striving to improve prediction accuracy and reliability. This includes creating more robust algorithms, refining data processing techniques, and developing better methods for interpreting model outputs. These advancements promise deeper insights into molecular systems and the creation of superior materials and technologies with enhanced performance characteristics.
Challenges and Future Directions of Quantum Chemistry Simulations in Drug Discovery
Computational Limitations and Resource Demands
Despite their power, quantum chemistry simulations frequently encounter substantial computational constraints. The complexity of molecular systems, particularly in drug discovery where large biomolecules interact with potential drugs, demands extraordinary computational power. This often results in protracted simulation times that may surpass current hardware capabilities. Researchers are actively pursuing innovative algorithms and approaches to address these limitations, including more efficient quantum algorithms and high-performance computing solutions.
Moreover, simulation accuracy often comes at the expense of computational efficiency. Striking the right balance is particularly challenging when dealing with complex drug candidates. Overcoming these resource-intensive obstacles is critical for broader implementation of quantum chemistry simulations in drug discovery pipelines.
Accuracy and Reliability of Quantum Chemical Methods
The precision of quantum chemical methods is paramount for trustworthy drug discovery predictions. Various methods offer different accuracy levels at varying computational costs. Selecting the optimal method for a specific system requires careful evaluation of these trade-offs. Ongoing research focuses on developing enhanced quantum chemical approaches that improve accuracy while reducing computational expenses.
Prediction reliability depends on multiple factors including input data quality, theoretical model selection, and approximation handling. Ensuring consistent, reproducible quantum chemistry results is fundamental for their practical application in pharmaceutical development.
Integration with Experimental Data and Biological Systems
Bridging quantum chemistry simulations with experimental data and biological systems is crucial for enhancing their predictive value. Validating simulation results against experimental findings from techniques like X-ray crystallography and NMR spectroscopy builds confidence in their accuracy. However, connecting quantum chemical insights to complex biological drug mechanisms remains challenging.
Developing robust frameworks to integrate simulations with experimental data and biological models is essential for a comprehensive understanding of drug-target interactions and improved drug design.
Development of Novel Drug Candidates and Design Strategies
Quantum chemistry simulations increasingly guide novel drug candidate design. By revealing molecular interactions and energy landscapes, these simulations help develop compounds with improved binding characteristics and specificity. They are particularly valuable for identifying critical molecular interactions and optimizing structures to enhance efficacy while minimizing toxicity.
These simulations also enable exploration of chemical space beyond traditional experimental limitations, potentially uncovering innovative drug candidates with superior therapeutic profiles.
Addressing the Challenges of Complexity and Scalability
The complexity of biological systems and the need for accurate predictions across diverse molecular interactions pose significant scalability challenges for quantum chemistry in drug discovery. Developing novel algorithms capable of handling complex systems—including large biomolecular assemblies and intricate reaction mechanisms—represents a critical research frontier.
Creating efficient computational frameworks to manage these high-dimensional systems is essential for wider adoption of quantum chemistry simulations in pharmaceutical research. This includes advancing theoretical models, improving algorithms, and leveraging state-of-the-art computing resources.